

The U.S. prescription drug market is highly dysfunctional, with high costs reducing use and thus leading to worse health outcomes among poorer Americans. In particular, the growing cost of biologic drugs—complex proteins derived from living organisms—threatens affordable care.
The lengthy patent monopolies sanctioned by Congress allow biologic manufacturers to raise prices ever higher. Patent monopolies–the exclusive rights to produce and market a drug–exist to reward pharmaceutical companies for the years of research and development and the many millions of dollars invested to create and test a drug through the approval process. But biologic manufacturers get even more time in the United States, in part, because language in the Affordable Care Act makes it harder to bring generic versions of biologic drugs–known as biosimilars–to market.
As our work at the Foundation for Research on Equal Opportunity has shown, delaying meaningful biosimilar competition by just a few years wastes billions of dollars in savings.
To help address this problem, Senators Mike Lee (R., Utah) and Ben Lujan (D., N.M.) have introduced the Biosimilar Red Tape Elimination Act. The bill recognizes that all rigorously tested biosimilars are as safe and effective as the reference biologics they mimic and are thus interchangeable. Europe has led the way for biosimilar drug approvals and it’s time for the United States to do the same.
For example, the European Union has already embraced biosimilars as interchangeable with their reference biologics. Last summer, the EMA issued a scientific statement on biosimilar interchangeability, noting that biosimilars “have been thoroughly reviewed and monitored over the past 15 years and the experience from clinical practice has shown that in terms of efficacy, safety and immunogenicity they are comparable to their reference products and are therefore interchangeable.”
Designating all biosimilars as interchangeable is one part of an array of legislative and regulatory changes that would significantly reduce the cost of biologics in the United States. Some researchers expect competition from biosimilars will save Americans $181 billion over the next five years, but these savings will only be realized if elected officials enact policies that foster a competitive market.
To reduce the cost of biologics as much as possible, the United States should reform its patent laws and address the unacceptably long exclusivity period granted by the FDA for biologics approvals. But changing existing law to deem all biosimilars as interchangeable would also have profound effects on the ability of biosimilar manufacturers to gain market share and thereby drive drug prices down.
Large drug companies oppose these reforms, but there is precedent for enacting them in the United States. Consider the success of low-cost generic versions of small molecule drugs. The Drug Price Competition and Patent Term Restoration Act of 1984, also known as Hatch-Waxman, facilitated the development and approval of generics, leading to greatly expanded drug access and lower costs to consumers.
One way that Hatch-Waxman encouraged generic drug development was through the Abbreviated New Drug Application (ANDA). To obtain FDA approval through an ANDA, the generic manufacturer must only prove that the generic works in the same manner as the original drug, rather than perform lengthy clinical trials to prove efficacy and safety among trial participants. As a byproduct of the ANDA, states began crafting laws allowing automatic substitution of generics at the pharmacy checkout counter. Automatic substitution helped fuel the market competition we see today by allowing pharmacists to dispense a generic version without getting permission from the doctor who prescribed the branded version.
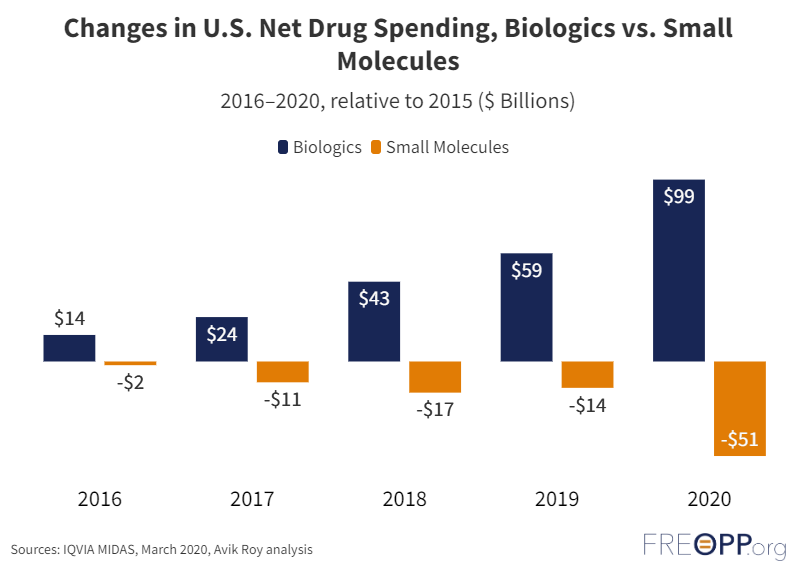
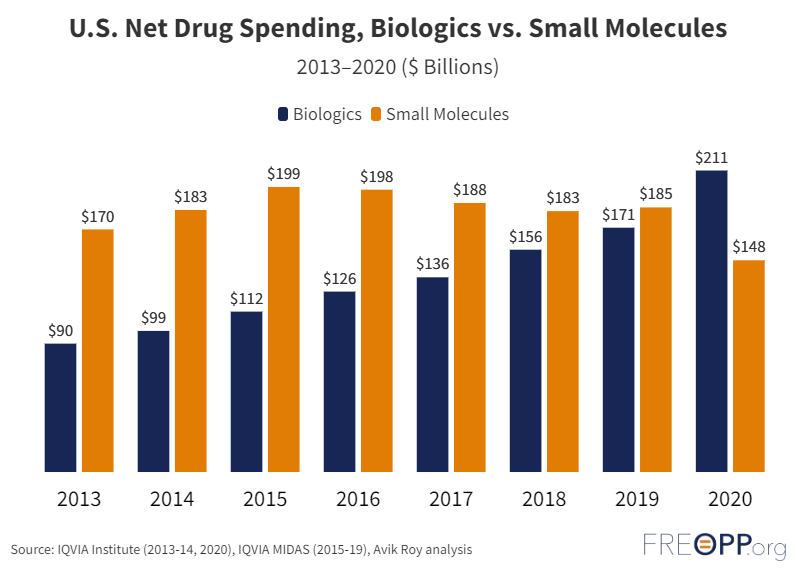
Thanks to Hatch-Waxman, 90 percent of drugs dispensed in the United States are generics: a rate higher than any other country. Furthermore, overall drug spending on small molecule drugs has decreased since 2016, despite increased prescribing volume; a clear indicator that prices for such drugs have fallen.
However, the ability to automatically substitute biosimilars for biologics is virtually nonexistent in the United States: only three U.S.-approved biosimilars have an interchangeability designation and are eligible for automatic substitution. This is because the development of biosimilars is regulated not by Hatch-Waxman, but by the Biologics Price Competition and Innovation Act (BPCI) as written in the Affordable Care Act. Under the reasoning that the complex nature of biologics necessitates more rigorous clinical testing, the BPCI requires biosimilar manufacturers to conduct switching studies to prove interchangeability. That is, trial participants are switched back and forth between the branded product and biosimilar to show both work equally. Switching studies add another costly and time-consuming barrier to market entry.
The barrier is worsened by doctors’ reluctance to prescribe biosimilars over original biologics. For example, a recent Cardinal Health Report showed that 70 percent of dermatologists, 65 percent of gastroenterologists, and 60 percent of rheumatologists considered designated interchangeability to be a very important consideration in prescribing any biosimilar in place of the popular drug Humira. By designating all biosimilars as interchangeable, physicians will have greater confidence in prescribing biosimilars to patients because the law will match what the science says.
To create a more efficient and cost-effective drug market, it is crucial to designate all biosimilars as interchangeable, extending the automatic substitution framework to biosimilars. This would promote competition, lower prices, and improve access to life-changing treatments. It is time for the United States to catch up to Europe's innovative approach and embrace biosimilars as interchangeable alternatives to biologics.
